帕金森小科普 - 概况
全球有大约 450 万帕金森病患者,近一半在中国。中国目前已有 220 万帕金森患者。我国 60 岁以上的老年人超过 1% 患有帕金森病,65 岁以上的老年人口中大约有 1.7% 的人患有帕金森病,70 岁以上患病率达 3%~5%,是继肿瘤、心脑血管病之后中老年的 「第三杀手」,而且每年新发病例近十万人。(来自网络)
帕金森小科普 - 发病机制及临床表现
帕金森病是除阿尔兹海默症之外的神经退行性疾病。临床上,该病的特征表现为动作迟缓、静止性震颤及僵硬,这是由于中脑腹侧黑质部分缺乏多巴胺能神经元所致(如下图 1)。
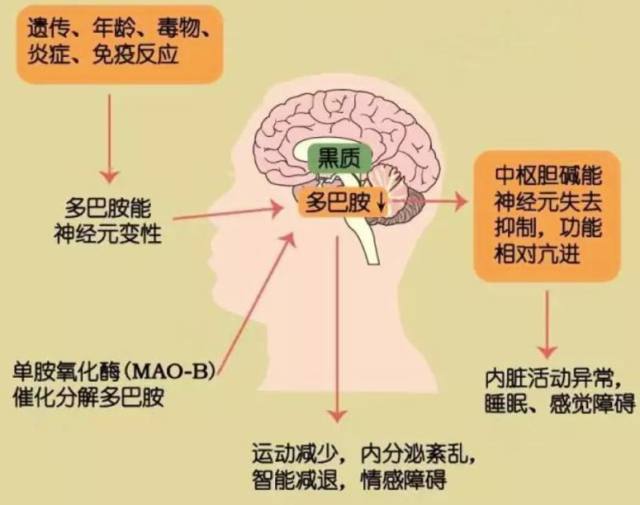
图 1 帕金森发病机制及临床表现
帕金森小科普 - 分子机制
正常情况下,突触前神经元中神经递质多巴胺的释放导致信号转导,通过 D1 和 D2 类型的多巴胺受体转导至突触后神经元。D1 受体的信号转导是通过 G 蛋白激活腺苷酸环化酶,形成环腺苷酸(cAMP)并激活 PKA 后完成的。D2 类型受体则通过抑制腺苷酸环化酶阻止此信号转导。(1-2)(如图 2, Normal State)。
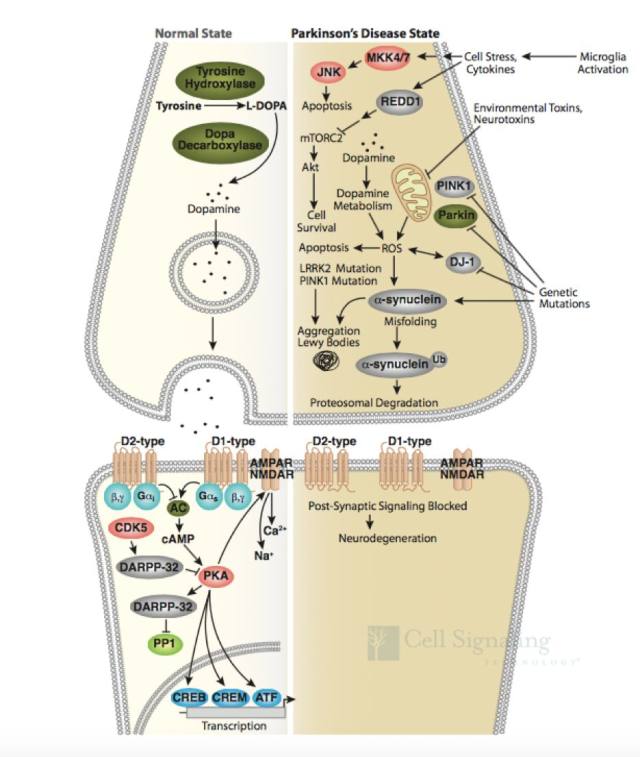
图 2 帕金森病信号通路
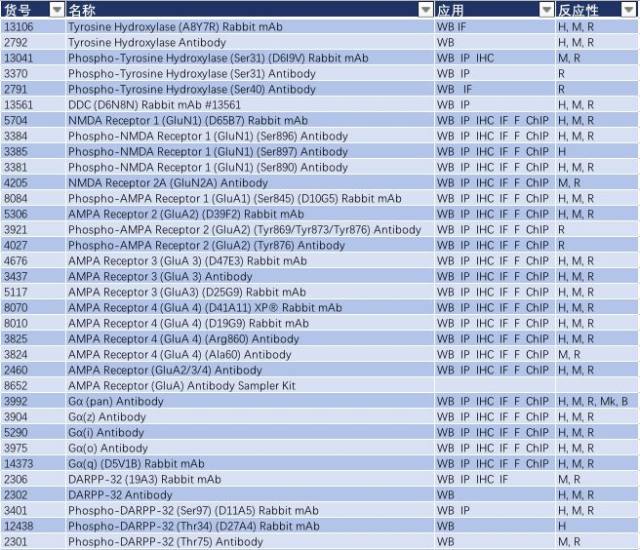
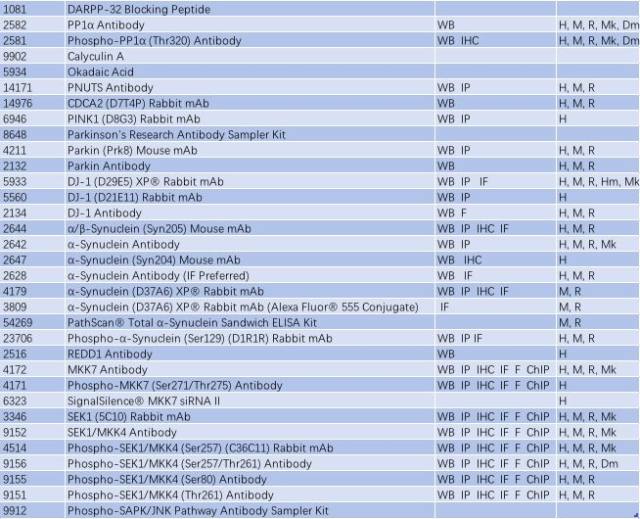
帕金森病可能是基因突变(家族性)和暴露于环境和神经毒素下(散发性)导致。Parkin、DJ-1 和 PINK1 发生功能丧失性突变和隐性遗传导致线粒体功能障碍和活性氧簇(ROS)积聚,而 α-突触核蛋白(α-synuclein)和 LRRK2 的错义突变和显性遗传可能影响蛋白降解通路,导致蛋白质聚集以及路易小体积聚(如图 2,Parkinsen’s Diease State),进而导致多巴胺能神经元过早退化或者导致多巴胺释放和多巴胺能神经传递受损,这可能是先于多巴胺能神经元坏死的早期病理变化前兆(3-5)。因此,研究帕金森的发病分子机制,Parkin、DJ-1 、PINK1、α-synuclein 和 LRRK2 这些经典的靶蛋白必不可少。CST 提供了这些靶蛋白的优质抗体,见表 1。
以上介绍是关于帕金森疾病的基础知识,我们最想知道的 「信号通路」 将会逐渐登场。说到相关信号通路,我们还要再温习一下帕金森病信号通路。从图 2 我们看到,帕金森病还有一个炎症部分,来源于小神经胶质细胞激活后导致炎症细胞因子释放和应激。小神经胶质细胞激活可通过 JNK 通路导致细胞凋亡(这里的细胞凋亡大多是受 NF-κB 调控的),并通过 REDD1 阻止 Akt 信号转导通路(如图 2,Parkinsen’s Diease State)。
表 1:
与帕金森相关的信号通路 - NF-kB
研究显示,炎症和免疫反应是帕金森疾病(PD)进展的决定性因素,也是遗传性和散发性 PD 发病的潜在因素之一。近来,研究报道的 PD marker 基因,如 SNCA (α-synuclein) 基因或 LRRK2(Leucine-rich repeat kinase 2), 通过激活小胶质细胞和星形胶质细胞刺激炎症反应直接参与 PD 的进展(6)。还有文章报道早期的 PD 患者在大脑黑质和硬核区存在激活的小胶质细胞(7)。由炎症信号通路的失调而引起小胶质细胞的激活都涉及到 PD 的神经炎症反应。激活的外周炎症有助于 PD 的发生和(或)进展,其与中枢炎症反应协同作用,促进使黑质致密部(SNpc)多巴胺能神经元变性(8-9)。
NF-κB,自发现至今已有 30 多年,研究人员已经证明 NF-κB 是各种促炎介质基因表达的「总开关」(10),它在神经系统的所有细胞中都表达,包括神经元、少突细胞、小胶质细胞、星形胶质细胞等(如图 3)(11,12,16)。NF-κB 通过调控促炎细胞因子 tumor necrosis factor (TNF)、白介素 1 (Interleukin 1, IL-1)、趋化因子 monocyte chemoattractant protein(MCP-1)等在炎症反应中起到非常重要的作用(16)。临床上用于应对炎症的氨基水杨酸盐及脂多糖等药物都是通过抑制 NF-κB 起作用的。同时,大量的研究表明,这些药物可以抑制小胶质细胞的激活,同时对多巴胺能神经元有一定的保护作用(13)。尽管 NF-κB 激活可以防止自激活的细胞凋亡,但是作为一个转录因子,它可调控细胞毒性药物如一氧化氮的产生,进而可能间接导致其他细胞的凋亡(图 3)。尤其是小胶质细胞,在激活的时候会产生神经毒性的活性氧等物质,这就解释了细胞因子引起的小胶质细胞激活是通过 NF-κB 激活(14)。脂多糖(LPS)能活化小胶质细胞,进而使 SNpc 部位多巴胺能神经元退化(15)。
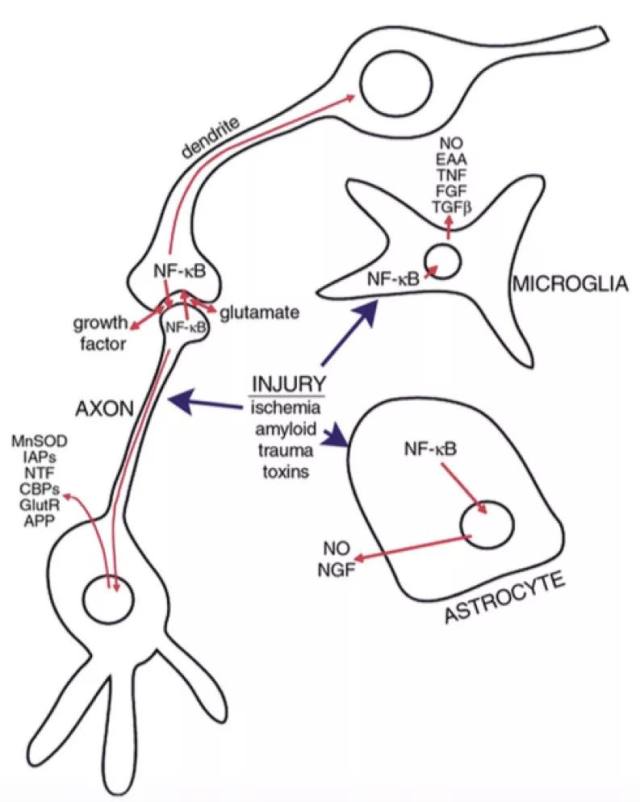
图 3 激活的 NF-κB 在神经元和胶质细胞中的复杂作用。
脑损伤后,神经元与胶质细胞中的 NF-κB 被激活。NF-κB 激活的神经元诱导调节突触可塑性的基因表达;NF-κB 激活的胶质细胞产生促炎细胞因子和潜在的神经毒性和刺激毒素(excitotoxins)(16)。
因此,NF-κB 信号通路的激活与慢性炎症反应密切相关,抑制小神经胶质细胞中 NF-κB 的活性可能是有效治疗 PD 的一个手段。研究 NF-κB 信号通路的激活可从以下靶点入手(表 2)。
表 2:
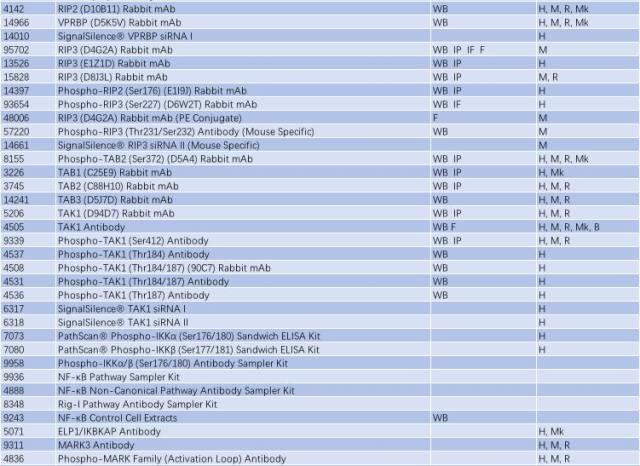
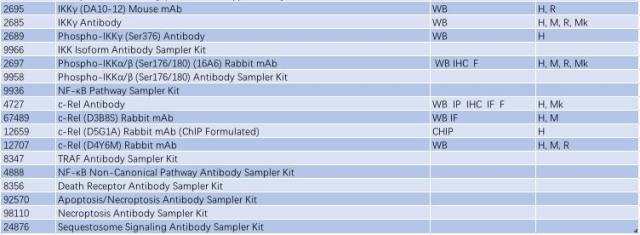
与帕金森相关的信号通路 - Jak/Stat
Jak/Stat 信号通路的激活对神经炎症及神经退行性疾病的影响是近几年中比较新的研究。Etty N.Benveniste 最近的研究报道了抑制 Jak/Stat 通路可以保护多巴胺神经元的退化(17)。
Etty 为了证明激活的 Jak/Stat 信号通路与帕金森的关系,分别在细胞水平和动物模型上做了深入的研究,文章数据详实、证据充分。通过 Jak1/2 抑制剂,AZD1480(该抑制剂可以减少 STAT1 和 STAT3 的激活)(如图 4,图中 P-STAT1 和 P-STAT3 的抗体都来自于 CST),抑制了小胶质细胞和巨噬细胞中的 MHCII 和炎症基因表达。在体内的研究中,研究者们使用了由 α-synuclein 病毒过表达的大鼠 PD 模型。AZD1480 通过抑制小胶质细胞活化、巨噬细胞和 CD4+ T - 细胞浸润和促炎细胞因子/趋化因子的产生,抑制了 α-synuclein 诱导的神经炎症。大量参与细胞信号、神经系统发育和功能、炎性疾病/过程,以及神经系统疾病的基因,在 α-synuclein 过表达的大鼠黑质中得到增强,并在 AZD1480 处理后受到抑制(图 5)。重要的是,体内实验还证明,抑制 JAK/Stat 通路的可抑制多巴胺能神经元变性(图 6)。
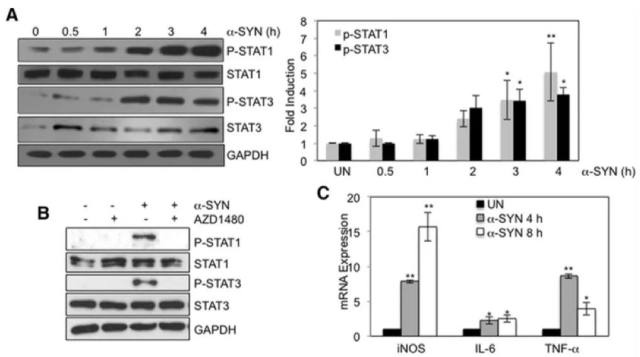
图 4 α-SYN 对 Stat 的激活及基因表达的作用
注:A. 小鼠 BMDMs 被体外合成的α-SYN (500 nM) 刺激至 4 h 后,STAT1/3 及其磷酸化基因的表达;B.BMDMs 在 AZD1480 (0.5 M) 的刺激 2 h 后,STAT1/3 磷酸化受到抑制;C. α-SYN 刺激引起炎症相关基因的表达

图 5 AZD1480 在体内抑制了α-SYN 激活的 Jak/Stat 信号通路
A.VH 或者 AZD1480 (10 mg/kg/d) 灌胃 AAV2-GFP 或 AAV2-SYN 感染 2 周的大鼠,2 周后取出黑质区,进行 RNA-seq;B. 聚类分析的热图,神经炎症等基因α-synuclein 过表达的大鼠黑质中得到增强,并在 AZD1480 处理后受到抑制。
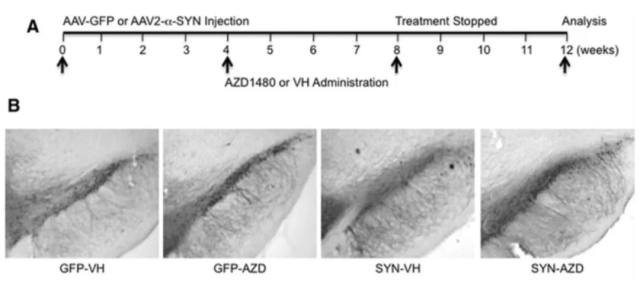
图 6 AZD1480 对黑质区 TH 阳性神经元的保护
A.AAV2- GFP 或 AAV2- SYN 感染大鼠 4 周和 VH 或者 AZD1480 (10 mg/kg/d) 处理进程;B.α-SYN 3 周内诱导了 50% 多巴胺能神经的减少,AZD1480 的处理使得这种情况减少。
这里只是把其中的一小部分图展示出来,若想看更详细的内容,请参考文章(17)。
此外,Piotr Przanowski 等报道了 Stat1 和 Stat3 诱导炎症相关基因 Jmjd3 的表达,以及促炎症细胞因子的产生,从而激活小胶质细胞。(18)。可见,Jak/Stat 信号通路也是通过一些神经炎症参与帕金森的进程。更多相关知识请见:https://www.cst-c.com.cn/contents/science/cst-pathways/science-pathways 及产品请看表 3。
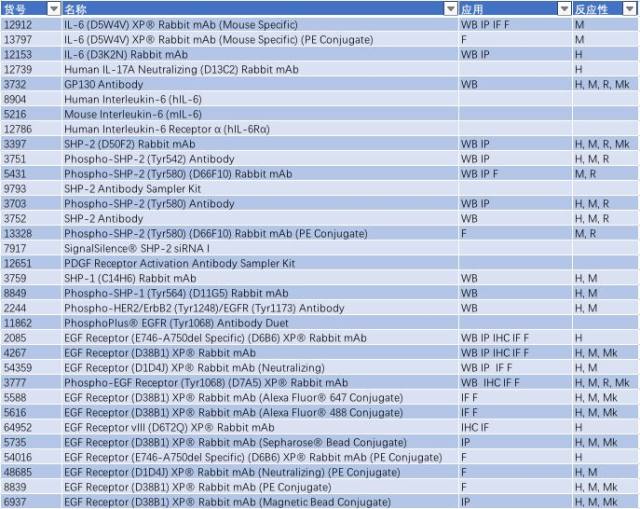
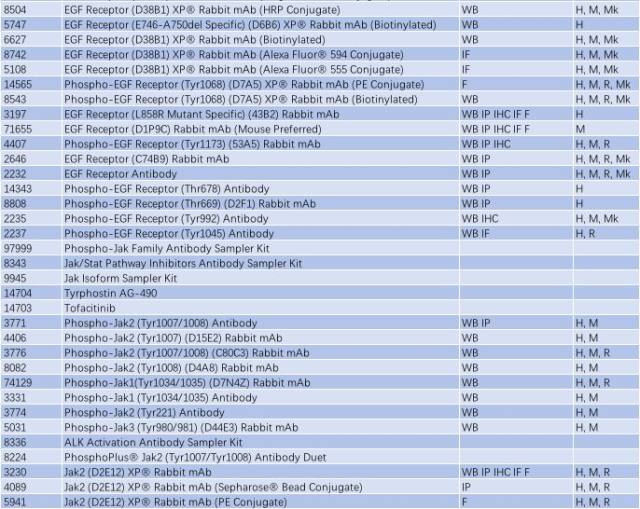
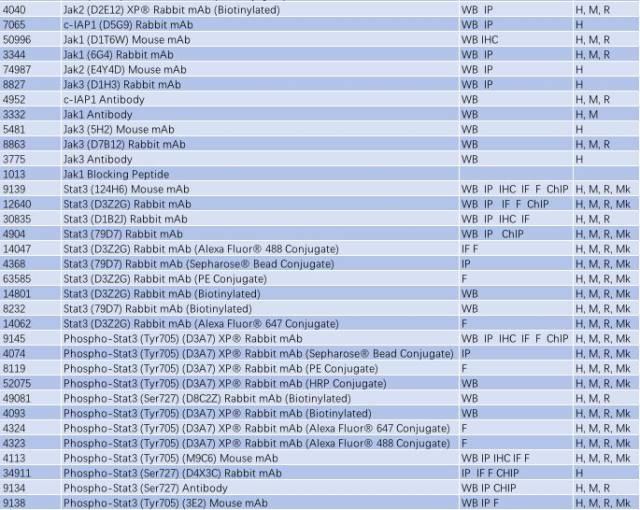
表 3:

与帕金森相关的信号通路 - TLR
Toll 样受体(TLRs)可识别不同的病原体相关分子模式,并在先天性免疫应答中扮演着不可或缺的角色。它们是抵御病原体入侵的第一道防线,在炎症、免疫细胞调控、存活和增殖方面发挥着关键作用。到目前为止,已发现 11 个 TLR 家族成员,其中 TLR1、TLR2、TLR4、TLR5、TLR6 和 TLR11 位于细胞表面,而 TLR3、TLR7、TLR8 和 TLR9 位于内体/溶酶体部分。TLR 信号转导通路的激活来源于细胞浆 Toll/IL-1 受体(TIR)的结构域,该结构域与 TIR 结构域包含的接头蛋白 MyD88 发生相互作用。经过配体的刺激,通过两个分子死亡结构域的相互作用,MyD88 将 IL-1 受体相关激酶-4(IRAK-4)吸引到 TLRs。IRAK-1 经磷酸化而激活并与 TRAF6 发生作用,从而激活 IKK 复合体并导致 NF-κB 的激活。激活的 NF-κB 被转移到细胞核中,又会促进各种各样的促炎分子的表达。从活化的小胶质细胞和局部微环境分泌的这些促炎分子增加了 SNpc 的氧化应激,从而导致多巴胺能神经元的退化(19-21)。
各种各样的促炎和神经毒性因子, 如过氧化物、TNF、IL 都是由激活的小胶质细胞分泌的。同时,这些促炎性因子促进小胶质细胞产生 mcp-1,MIP-1,而这些蛋白也反过来影响神经炎症的发生,早期的细胞因子是通过与 Toll-like receptor (TLR) 4 结合而发挥作用。当这些细胞因子释放出来,进而诱导 Stat1 和 Stat3 的激活(22)。
可见,TLR 信号通路与 NF-κB 和 Jak/Stat 信号通路紧密连接参与到帕金森病的发生及发展。更多细节,可以查看 TLR 信号通路。相关产品见表 4。
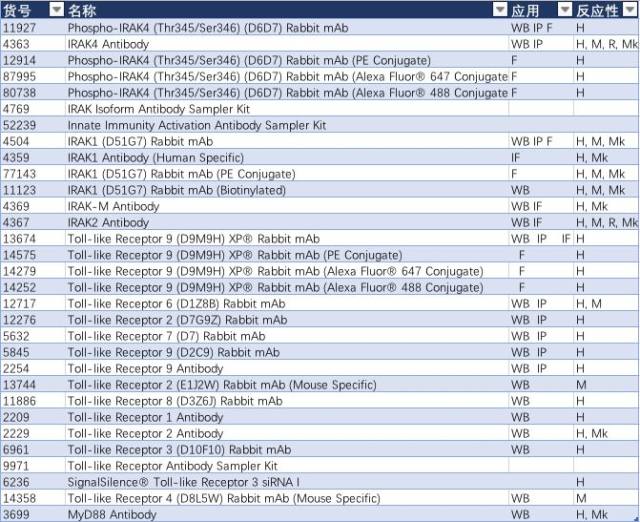
表 4:
结语
总之,我们可以用一张图来总结参与到帕金森病中的各神经炎症信号通路。
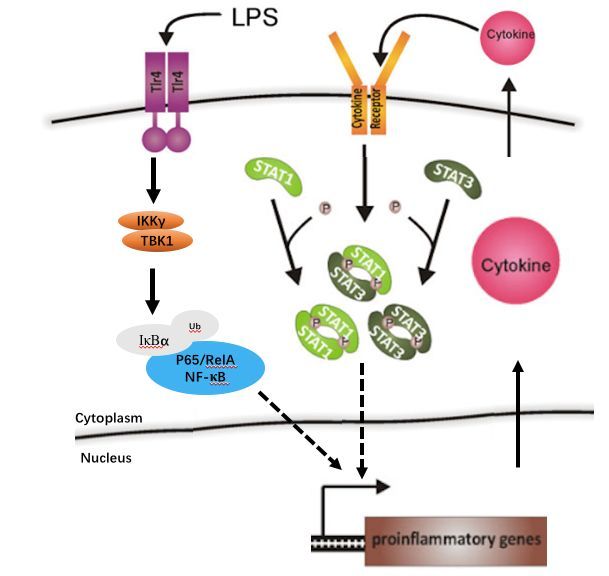
若您喜欢这篇文章,请快快收藏吧,说不定哪天就用上了。
参考文献:
1. Dauer W, Przedborski S.etal. Parkinson'sdisease: mechanisms and models. Neuron.2003;39(6), 889–909.
2. Girault JA, Greengard P.et al Theneurobiology of dopamine signaling. Arch Neurol. 2004;61(5), 641–4.
3. Imai Y, Lu B ,et al. Mitochondrialdynamics and mitophagy in Parkinson's disease: disordered cellular power plantbecomes a big deal in a major movement disorder. Curr Opin Neurobiol. 2011;21(6), 935–41.
4. Patten DA, Germain M,Kelly MA, Slack RS. Reactiveoxygen species: stuck in the middle of neurodegeneration. J. Alzheimers Dis. 2010;20 Suppl 2, S357–67.
5. Springer W, Kahle PJ .Regulationof PINK1-Parkin-mediated mitophagy. Autophagy.2011;7(3), 266–78.
6. Barcia C. Glial-mediated inflammation underlying parkinsonism. Scientifica (Cairo). 2013;357805.
7. Dzamko N, Geczy CL, Halliday GM. Inflammation isgenetically implicated in Parkinson’s disease. Neuroscience. 2015;302:89-102.
8. Bove J, Perier C. Neurotoxin-based models ofParkinson’s disease. Neuroscience. 2012;211:51-76.
9. Kanaan NM, Kordower JH, Collier TJ. Age-relatedchanges in glial cells of dopamine midbrain subregions in rhesus monkeys. Neurobiol Ag- ing. 2010;31(6):937-952.
10. TsoulfasG, GellerDA.NF-κBintransplantation:friendorfoe?Transpl Infect Dis. 2001;3(4):212-219.
11. Baeuerle, P., and Baltimore, D. NF-κB: ten yearsafter. Cell. 1996; 87:13–20.
12. O』Neill, L.A., and Kaltschmidt, C. NF-kappa B: acrucial tran- scription factor for glial and neuronal cell function. Trends Neurosci. 1997; 20:252–258.
13. Roebuck KA, Carpenter LR, Lakshminarayanan V,Page SM, Moy JN, Thomas LL. Stimulus-specific regulation of chemokineexpression involves differential activation of the redox-responsivetranscription factors AP-1 and NF-κB. JLeukoc Biol. 1999;65(3):291-298.
15. Liu M, Bing G.Lipopolysaccharide animal models for Parkinson’s disease. Parkinsons Dis. 2011;2011:327089.
16. NF-κB in neuronal plasticity and neurodegenerative disorders. Journal of ClinicalInvestigation, 2001;107(3), 247-254
17. Hongwei Qin,JessicaA. Buckley ,et al.Inhibition of the JAK/STAT PathwayProtects Against Synuclein-Induced Neuroinflammation and Dopaminergic . The Journal of Neuroscience, 2016 ;36(18):5144 –5159
18. Przanowski P, Dabrowski M, Ellert-Miklaszewska A, et al. Thesignal transducers Stat1 and Stat3 and their novel target Jmjd3 drive theexpression of inflammatory genes in microglia. J Mol Med (Berl). 2014;92(3):239- 254
19. Akira S, TakedaK. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499-511.
20. Banerjee A, Gerondakis S. Coordinating TLR-activated signalingpathways in cells of the immune system. Immunol Cell Biol. 2007;85(6):420-424.
21. Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in theMyD88-independent Toll-like receptor signaling pathway. Science. 2003;301(5633):640-643.
22. Tanaka S, IshiiA, Ohtaki H, Shioda S, Yoshida T, Numazawa S. Activa- tion of microglia inducessymptoms of Parkinson’s disease in wild-type, but not in IL-1 knockout mice. JNeuroinflammation. 2013;10:143
本文来自「CST博士互助平台」,更多相关内容,
请关注👇

点击此处,阅读原文。